🧮 计算器脚本
计算器模块用于从 MD 轨迹、NEP 模型和结构文件中计算材料性质。
脚本位置: Scripts/calculators/
功能概览
计算器模块大致可以分为三类:
- 轨迹性质: 从 GPUMD 或 extxyz 轨迹计算 MSD、离子电导率等时间相关性质;
- NEP 辅助计算: 使用 NEP 模型预测能量/力/应力、提取描述符、计算 DOAS、进行 NEB 或结构优化;
- 极性材料分析: 为钙钛矿和极性材料构建近邻列表、计算局域位移、平均结构、八面体倾转和局域极化。
如果你还不熟悉某个脚本,建议先用交互模式查看输入提示;如果已经知道参数,则可以直接使用 gpumdkit.sh -calc ... 调用。
| 任务 | 命令 | 主要输入 |
|---|---|---|
| 离子电导率 | gpumdkit.sh -calc ionic-cond <element> <charge> |
msd.out、thermo.out、model.xyz |
| MSD | gpumdkit.sh -calc msd <trajectory.xyz> <element> <dt_fs> |
extxyz 轨迹 |
| NEP 预测 | gpumdkit.sh -calc nep <input.xyz> <output.xyz> <nep.txt> |
extxyz + NEP 模型 |
| NEP 描述符 | gpumdkit.sh -calc des <input.xyz> <output.npy> <nep.txt> <element> |
extxyz + NEP 模型 |
| DOAS | gpumdkit.sh -calc doas <input.xyz> <nep.txt> <output.txt> |
extxyz + NEP 模型 |
| NEB | gpumdkit.sh -calc neb <initial.xyz> <final.xyz> <n_images> <nep.txt> |
初末态结构 |
| 结构优化 | gpumdkit.sh -calc minimize <structure> <nep.txt> [fmax] [max_steps] |
结构 + NEP 模型 |
| 邻居列表 | gpumdkit.sh -calc nlist ... |
参见 极性材料分析 |
| 位移 | gpumdkit.sh -calc disp ... |
参见 极性材料分析 |
| 平均结构 | gpumdkit.sh -calc avg-struct ... |
参见 极性材料分析 |
| 八面体倾转 | gpumdkit.sh -calc oct-tilt ... |
参见 极性材料分析 |
| ABO3 极化 | gpumdkit.sh -calc pol-abo3 ... |
参见 极性材料分析 |
查看完整帮助:
命令行帮助表类似:
+-------------------------------------------------------------------------------------------------------+
| CALCULATOR TOOLS |
+-------------------------------------------------------------------------------------------------------+
| Usage: gpumdkit.sh -calc <type> [args...] |
+-------------------------------------------------------------------------------------------------------+
| ionic-cond <element> <charge> Calculate ionic conductivity from MSD data |
| nep <input.xyz> <output.xyz> <nep_model> Calculate energy/force/virial with a NEP model |
| des <input.xyz> <output.npy> <nep_model> <el> Calculate NEP descriptors for one element |
| doas <input.xyz> <nep_model> <output.txt> Calculate density of atomistic states |
| neb <initial.xyz> <final.xyz> <n_images> <nep> Run NEB calculation with a NEP model |
| minimize <structure> <nep_model> [fmax] [n] Minimize a structure with a NEP model |
| msd <trajectory.xyz> <element> <dt_fs> [n] Calculate MSD from an extxyz trajectory |
| nlist [script args...] Build neighbor lists |
| disp [script args...] Calculate displacement from trajectory |
| avg-struct [script args...] Calculate averaged structure |
| oct-tilt [script args...] Calculate octahedral tilt |
| pol-abo3 [script args...] Calculate local polarization for ABO3 |
+-------------------------------------------------------------------------------------------------------+
交互模式中选择 4) Calculators,菜单如下:
+----------------------------------------------------------+
| CALCULATOR TOOLS |
+----------------------------------------------------------+
| 401) Calc ionic conductivity |
| 402) Calc properties by nep |
| 403) Calc descriptors of specific elements |
| 404) Calc density of atomistic states (DOAS) |
| 405) Calc nudged elastic band (NEB) by nep |
| 406) Build neighbor list |
| 407) Calc displacement from trajectory |
| 408) Calc averaged structure |
| 409) Calc octahedral tilt |
| 410) Calc polarization for ABO3 |
| 411) Minimize structure by nep |
| 412) Calc mean square displacement (MSD) from trajectory |
+----------------------------------------------------------+
| 000) Return to the main menu |
+----------------------------------------------------------+
Input the function number:
离子电导率
calc_ion_conductivity.py 从 msd.out 计算离子扩散系数和离子电导率。
交互模式选择 401,会看到:
>-------------------------------------------------<
| This function calls the script in calculators |
| Script: calc_ion_conductivity.py |
| Developer: Zihan YAN (yanzihan@westlake.edu.cn) |
>-------------------------------------------------<
Input <element> <charge> (eg. Li 1)
------------>>
常用输入文件:
| 文件 | 作用 |
|---|---|
msd.out |
必需,MSD 数据 |
thermo.out |
可选,自动读取温度 |
model.xyz |
可选,自动读取体积和离子数 |
run.in |
可选,识别扩胞信息 |
如果缺少自动识别所需文件,脚本会交互式询问温度、体积和离子数。
手动模式的提示类似:
Files 'thermo.out' and 'model.xyz' are not found.
Please provide the following values:
--------------------------->
Enter average temperature (in K):
Enter system volume (in A^3):
Enter number of ions:
输出示例:
Diffusivity (D):
D_x: 4.153e-07 cm^2/s
D_y: 4.174e-07 cm^2/s
D_z: 2.610e-07 cm^2/s
D_total: 3.646e-07 cm^2/s
------------------------------
Ionic Conductivity:
Sigma_x: 2.576e-02 mS/cm
Sigma_y: 2.589e-02 mS/cm
Sigma_z: 1.619e-02 mS/cm
Sigma_total: 2.261e-02 mS/cm
从轨迹计算 MSD
calc_msd.py 可以直接从 extxyz 轨迹中计算指定元素的 MSD。
交互模式选择 412,会看到:
Input <extxyz_file> <element_symbol> <dt_fs> [max_corr_steps]
Optional argument: max_corr_steps (default: frame number)
Example: dump.xyz Li 10
------------>>
参数含义:
| 参数 | 含义 |
|---|---|
dump.xyz |
输入轨迹 |
Li |
目标扩散元素 |
10 |
帧间隔,单位 fs |
5000 |
可选,最大相关步数 |
输出:
msd.out
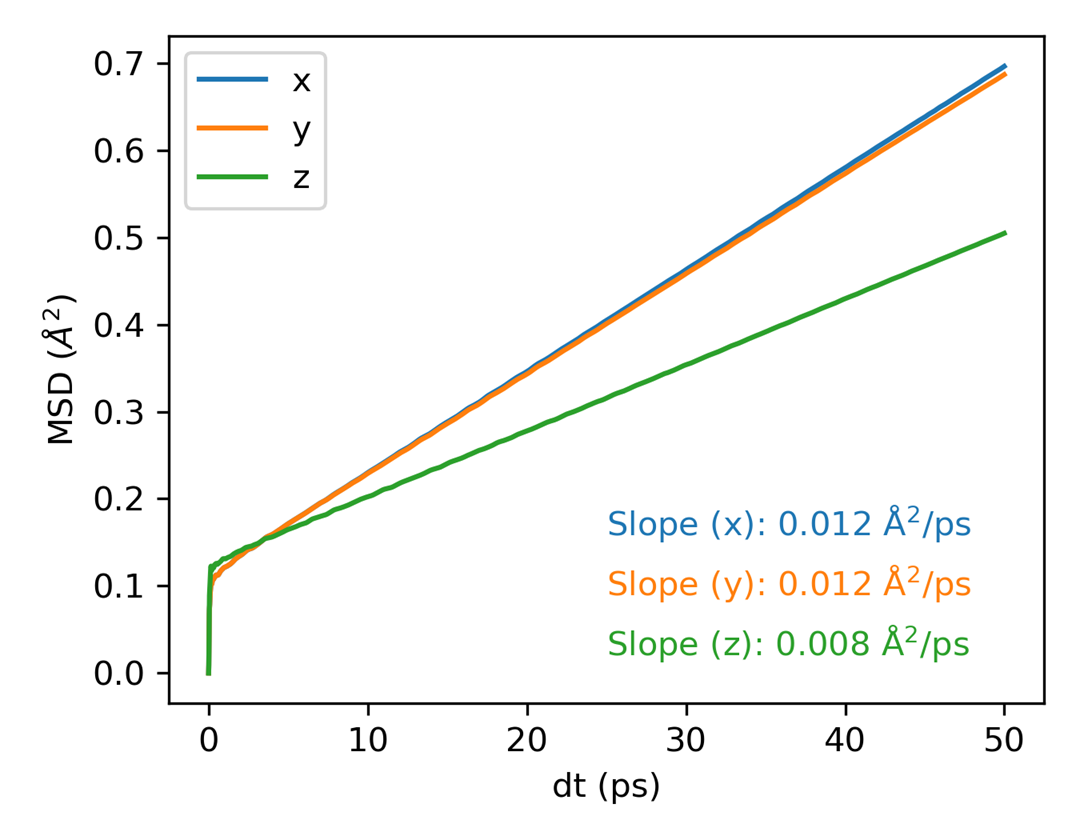
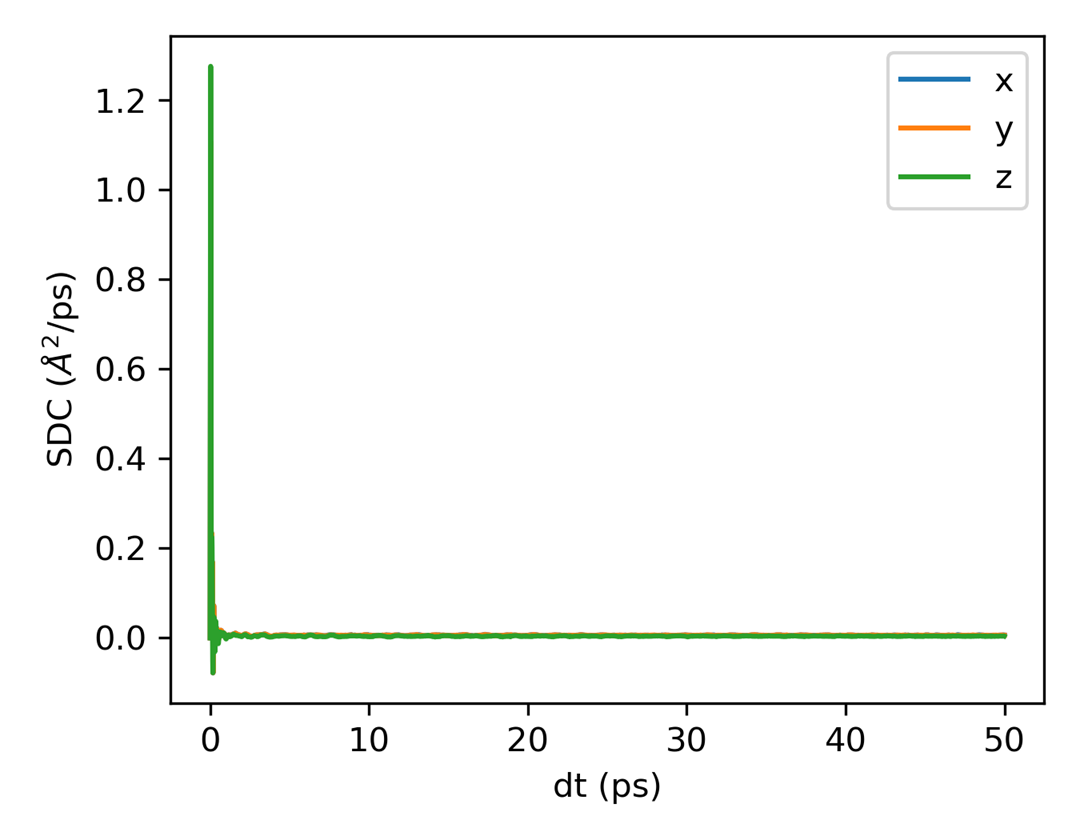
msd.out 的开头是文本表格,包含时间和不同方向的 MSD 列。生成后可以继续使用绘图命令检查扩散区间和自扩散系数。
随后可绘图:
NEP 预测性质
calc_properties_with_nep.py 使用训练好的 NEP 模型计算能量、力和应力。
依赖: calorine
使用此功能可以用训练好的 NEP 模型进行预测。建议在使用前先验证模型在目标结构上的预测质量,再将输出作为后续分析或筛选依据。
提示: 预测前可以清理 extxyz 元数据:
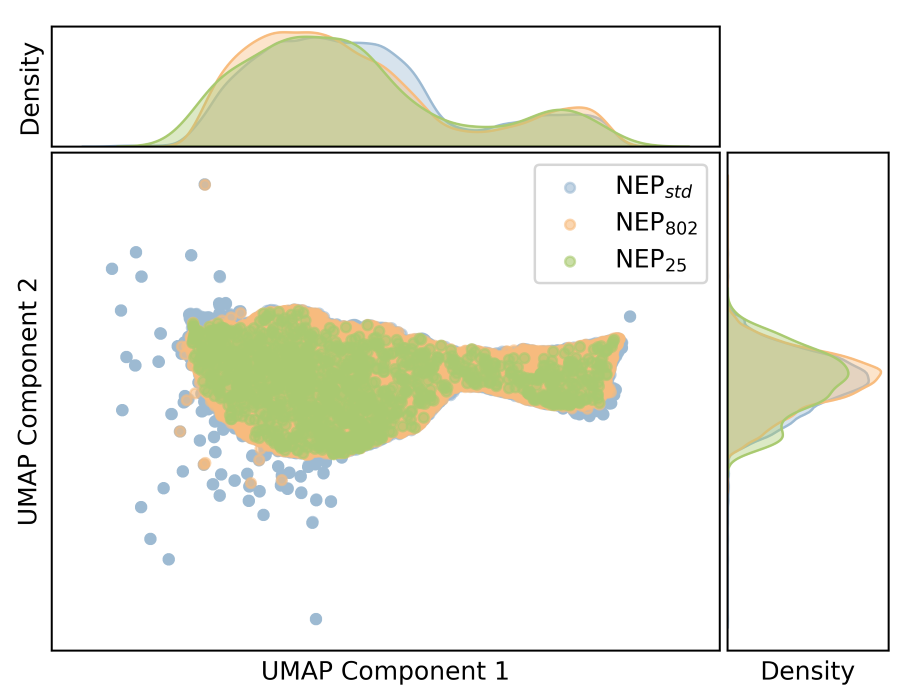
NEP 描述符
用途包括:
- 用 PCA/UMAP 可视化局域环境;
- 比较训练集和候选结构;
- 判断新数据是否扩展了描述符空间。
绘图:
DOAS
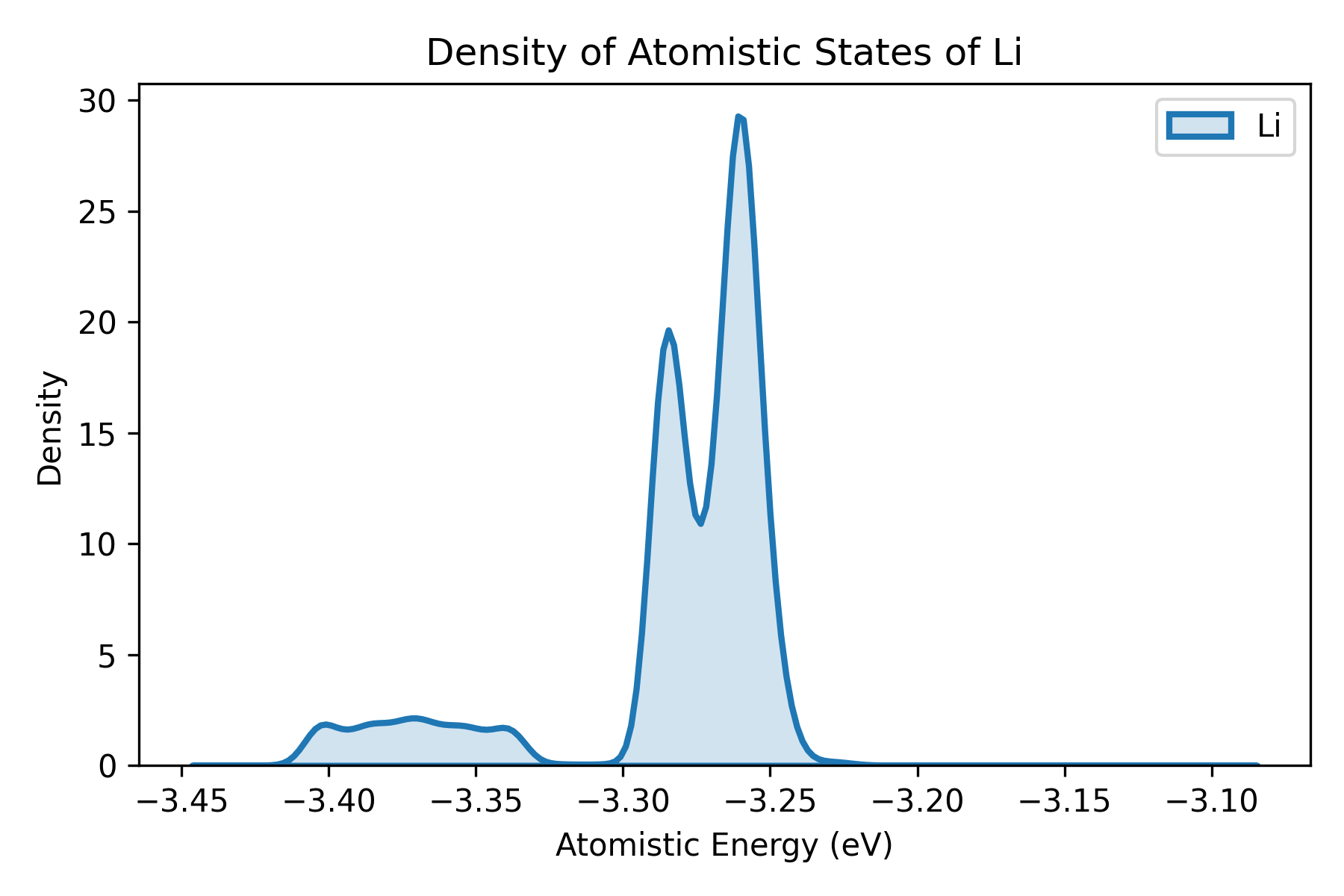
calc_doas.py 用于计算 Wang et al. 提出的 density of atomistic states。
脚本会:
- 读取所有结构;
- 使用 NEP 进行结构优化;
- 提取每个原子的能量;
- 按元素分组输出;
- 用绘图脚本可视化分布。
对于很大的体系,如果主要目的是获得优化后的结构和原子能量分布,直接在 GPUMD 中完成最小化和原子能量输出通常会更高效。
NEB 和结构优化
NEB:
这会使用 NEP 模型运行含 9 个中间像的 NEB 计算。运行过程中脚本会询问原子固定方式:
none:不固定原子;index:按原子序号固定;element:固定某一种元素的所有原子;position:固定坐标范围内的原子。
结构优化(通过 calorine):
依赖: calorine
输出包括:
minimize.xyzminimize.log
使用 OVITO 计算 RDF
rdf_calculator_ovito.py 使用 OVITO 的分析工具计算径向分布函数。
依赖: OVITO
输入文件: 结构文件(单帧或轨迹)
参数:
| 参数 | 含义 |
|---|---|
trajectory.xyz |
输入结构文件 |
6.0 |
RDF 计算的最大距离(Å) |
400 |
直方图的分箱数 |
可视化:
注意: 建议优先使用 GPUMD 中的 compute_rdf 命令。
铁电和极性材料工具
选项 406–410 面向钙钛矿和极性材料分析,常用于从 MD 轨迹中提取局域结构信息,然后进一步分析相变、畴结构或极化纹理。
这些脚本在本页只做快速索引,因为它们通常需要组合使用。推荐阅读专题教程中的完整工作流和参数说明:极性材料分析。
| 菜单 | CLI 子命令 | 作用 | 详细说明 |
|---|---|---|---|
406 |
gpumdkit.sh -calc nlist ... |
构建指定中心原子和近邻原子之间的近邻列表 | 极性材料分析 |
407 |
gpumdkit.sh -calc disp ... |
根据轨迹和近邻列表计算局域位移 | 极性材料分析 |
408 |
gpumdkit.sh -calc avg-struct ... |
从轨迹中计算平均结构 | 极性材料分析 |
409 |
gpumdkit.sh -calc oct-tilt ... |
计算八面体倾转角 | 极性材料分析 |
410 |
gpumdkit.sh -calc pol-abo3 ... |
根据 Born 有效电荷估算 ABO3 局域极化 | 极性材料分析 |
部分脚本需要安装 ferrodispcalc:
一个典型顺序是先用 nlist 建立近邻列表,再用该近邻列表计算位移、倾转或极化。对于平均结构,可以直接从轨迹出发,用长度容差控制哪些帧参与平均。